

Muskeldystrophie, progressive
Synonyme
Dystrophia muscularis progressiva
Englischer Begriff
Progressive muscular dystrophy
Definition
Fortschreitende genetisch bestimmte, primär degenerative Muskelerkrankungen, die einerseits eine klinisch und genetisch heterogene Krankheitsgruppe darstellen, andererseits sich aber durch die klinischen und laborchemischen Befunde von anderen Muskelerkrankungen abgrenzen lassen.
Pathogenese
Den Muskeldystrophien liegen Genmutationen zugrunde. Bei den Dystrophino- und Sarkoglykanopathien sind diese bekannt, ebenso bei den Calpaino-und α2-Laminopathien. Es gelingt nicht immer, Geno- und Phänotyp einander sicher zuzuordnen. Bei den Dystrophino-und Sarkoglykanopathien besteht ein Mangel an Strukturproteinen. Die Zusammengehörigkeit der Krankheitsgruppe zeigt sich in den histologischen Befunden. Alle Dystrophieformen gehen mit Degeneration und Umbau von Muskelgewebe, einschließlich des Sarkolemms einher. Es kommt zu Fibrosierungen und Fettzelleinlagerungen. Daneben finden sich auch regenerierende Muskelfasern. Der Muskelfaserdurchmesser variiert durch Hypertrophie und Atrophie von Muskelfasern stark. Es finden sich zentralständige Kerne und zelluläre Infiltrate. Sekundär kommt es zum Untergang von Strukturproteinen (Nebulin, Titin), was die Stabilität der Zelle beeinflusst. Auch im Stoffwechsel der Muskelzellen finden sich Störungen, die Glukoseutilisation ist herabgesetzt, Kreatinurie ist vermehrt bei verminderter Kreatininausscheidung und die 3-Methylhistidin- und Carnitinausscheidung sind vermehrt. Auch muskelspezifische Enzyme sind im Plasma erhöht.
Symptome
Abbildung 1 gibt eine Übersicht über die wichtigsten Muskeldystrophien und deren spezifische Symptome. Im Allgemeinen kommt es bei den Muskeldystrophien bei Kindern zu einer Entwicklungsverzögerung, rasche Ermüdbarkeit, Stolpern und Stürze, schwächebedingte Ungeschicklichkeit, herabgesetzter Muskeltonus, Muskelatrophie, Muskelschmerzen und Muskelkrämpfe. Häufig können eine Instabilität des Oberkörpers und ein Schaukeln in der Hüfte beobachtet werden.
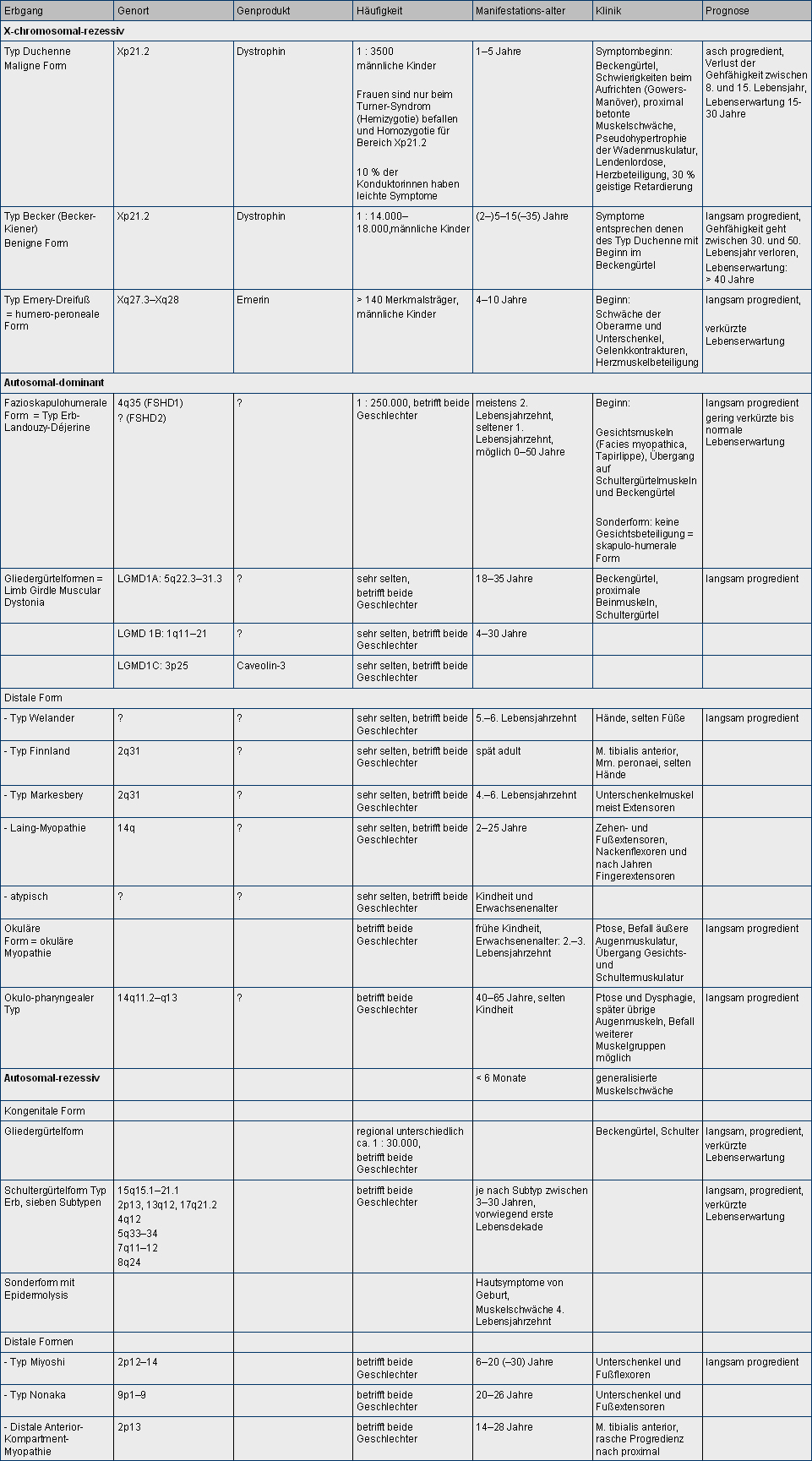
Abb. 1.
Klassifikation der Muskeldystrophien.
Diagnostik
Anamnese, Familienanamnese; Labor: Serumenzyme wie CK, GOT, GPT, LDH; genetische Tests; motorische und sensible Neurographie; Elektromyographie: neuromuskuläre Übertragung (repetitive Nervenstimulation); Muskelbiopsie: Histologie, Histochemie, Enzymhistochemie; Immunhistologie mit mono- und polyklonalen Antikörpern; Elektronenmikroskopie; Biochemie; Bildgebung: Muskelsonographie, Magnetresonanztomographie.
Differenzialdiagnose
Muskeldystrophie Typ Duchenne: kongenitale Myopathie, Merosinopathie (autosomal-rezessiv), chronische Form der Polymyositis, Dystrophia myotonica.
Muskeldystrophie Typ Becker: Polymyositis, spinale Muskelatrophie Kugelberg-Welander.
Therapie
Keine spezifische Therapie.
Konservative/symptomatische Therapie
Physiotherapie zum Erhalt der Gehfähigkeit.
Medikamentöse Therapie
Experimentell: Kortikosteroide. Therapiestudien: Gentamycin beeinflusst Stop-Codon, im Tierexperiment erfolgreich, in klinischen Studien bisher Creatinkinaseabnahme, keine Zunahme des Dystrophins. Weitere Studien werden mit Albuterol, Oxandrolone, Coenzym Q, Prednisolon, Creatine durchgeführt. Bisher hat keine der Studien, soweit analysiert, signifikante Verbesserungen erbracht.
Neurotrophische Faktoren.
Operative Therapie
Korrektur von Gelenkkontrakturen und Wirbelsäulendeformitäten. Experimentell: Plasmidinjektion; Transplantation von Muskelzellen und Muskel-Precursor-Zellen zur Freisetzung von Dystrophin; Problem: Abstoßungsreaktion, Überleben der transplantierten Zellen.
Dauertherapie
Siehe oben Physio-und Ergotherapie.
Prophylaktische Maßnahmen: Bei Narkosen ist Vorsicht geboten, Halothan und Succinylcholin sollten wegen der Gefahr der Rhabdomyolyse vermieden werden. Triggerfreie Narkosen müssen wegen Gefahr der malignen Hyperthermie angewandt werden.
Bewertung
Siehe Abbildung 1.
Autor
Iris Reuter
Anzeige

© Springer 2017 |
Powered by kb-soft |
|