
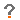

Amyotrophische Lateralsklerose
Synonyme
Myatrophische Lateralsklerose; Amyotrophe Lateralsklerose; ALS; Charcot-Krankheit; Motoneuronenerkrankung
Englischer Begriff
Motoneuron disease; Amyotrophic lateral sclerosis
Definition
Progressive neurodegenerative Erkrankung des 1. und 2. Motoneurons, die mit peripheren und zentralen Lähmungen einhergeht.
Pathogenese
Die Erkrankungshäufigkeit betrifft 6–8 : 100.000 Einwohner mit einer Inzidenz von 1,5–2 Erkrankungen pro 100.000 Einwohner pro Jahr (Männer : Frauen: 1,2–2,6 : 1). Die amyotrophe Lateralsklerose zählt zu den degenerativen Motoneuronenerkrankungen. Sie ist zu 5–10 % erblich (autosomal-dominanter Erbgang), der Rest ist sporadisch. 20 % der Patienten zeigen einen Defekt auf Chromosom 21 im Gen für die Superoxiddismutase 1 (SOD 1). Das Erkrankungsalter der erblichen Erkrankungen ist niedriger.
Es findet sich eine Degeneration der Motoneurone im Bereich der Vorderhörner, des Rückenmarks, der Hirnrinde und der Hirnnervenkerne. In den betroffenen Zellen werden Einschlusskörperchen gefunden. Besonderes Interesse gilt den ubiquitinpositiven Einschlüssen in den Vorderhornzellen. Ausgenommen sind die okulomotorischen Hirnnerven und der Onuf-Kern (Nucleus nervi pudendi) für den Analsphinkter.
Symptome
Häufig ist das erste Zeichen eine einseitig einsetzende distale periphere Lähmung, 20–30 % der Patienten, insbesondere ältere Frauen, haben eine bulbäre oder pseudobulbäre Symptomatik. Später liegen periphere und zentrale Lähmungen nebeneinander vor. Muskelatrophien und Faszikulationen zeigen die Degeneration des 2. Motoneurons an, ebenso Muskelkrämpfe, welche zusammen mit einer Muskeltonuserhöhung, lebhaften Reflexen und Pyramidenbahnzeichen auftreten (Degeneration des 1. Motoneurons). Im Allgemeinen sind die kognitiven Funktionen, Sensibilität, Sphinkterfunktionen intakt, jedoch können leichte sensible Störungen vorkommen. Die Ateminsuffizienz tritt im Endstadium dazu.
Die kognitiven Leistungen sind im Allgemeinen ungestört, es gibt jedoch eine familiäre Art der ALS, welche mit einer Demenz assoziiert ist. Die Demenz ist vom frontotemporalen Typ, der Genlocus hierfür ist das Chromosom 9.
Diagnostik
Kernspintomographie der Hals- und Brustwirbelsäule zum Ausschluss einer Myelopathie; Labor: Schilddrüsenserologie, Immunelektrophorese, Gamma-Globuline, GM1-Antikörper, Nieren- und Leberwerte, Blutbild, Lumbalpunktion zum Ausschluss eines infektiösen Geschehens; Elektromyographie: Nervenleitgeschwindigkeit, Riesenpotentiale.
Diagnosekriterien nach El Escorial siehe Abb. 1.

Abb. 1.
Diagnosekriterien nach El Escorial (Borasio 2003).
Differenzialdiagnose
Myelopathie, mu.jpgokal motorische Neuropathie, multiple Sklerose, Myopathie, HIV, Lues, Neuroborreliose, Postpoliosyndrom, paraneoplastische Syndrome (z. B. ALS bei Mammakarzinom), spinale Muskelatrophien.
Therapie
Konservative/symptomatische Therapie
Angehörigenschulung, Aufklärung der Angehörigen, Stützung des Patienten, Physiotherapie, Schlucktraining bei Dysphagie, logopädisches Training bei Dysarthrie, nicht-invasive Maskenbeatmung bei Dyspnoe, Erlernen von Kommunikation über Schreibmaschine oder Computer.
Medikamentöse Therapie
Riluzol (Rilutek) als antiglutamaterge Substanz, Verlängerung der Lebenserwartung für ca. drei Monate. Die Krankheitprogredienz wird nicht wesentlich reduziert, jedoch die Zeit bis zur Ateminsuffizienz verlängert.
Symptomatisch: Mestinon hilft bisweilen der Muskelkraft beim Schlucken.
Experimentell: Gabe von CNTF, BDNF und IGF1 ohne sichere Wirkung.
Dauertherapie
Rilutek
Bewertung
Im Allgemeinen rasche Progredienz; 10 % der Patienten überleben 10 Jahre; bei initial bulbärer Symptomatik ungünstige Prognose: 2–2,5 Jahre Überlebenszeit; übrige Patienten drei bis fünf Jahre. Rarität: Überlebenszeit über 40 Jahre.
Junges Erkrankungsalter und primäre Lateralsklerose (Degeneration der Pyramidenbahn mit weniger starken bulbären Symptomen) zeigen eine günstige Erkrankung mit langsam progredientem Verlauf an.
Nachsorge
Regelmäßige Kontrolluntersuchungen zur Anpassung der Therapie.
Autor
Iris Reuter
Anzeige

© Springer 2017 |
Powered by kb-soft |
|